
Antiviral Innate Immunity
To efficiently replicate within a host cell viruses have to circumvent intrinsic immune barriers. This includes avoiding recognition (sensing) by specific receptors and counteracting or evading antiviral inhibitory mechanisms within the cell. The research of our laboratory focuses on the innate immune response to viral infections and mobile genetic elements. We are interested in understanding how intrinsic immune factors sense, signal, and block the replication of retroviruses as well as other DNA- and RNA-viruses and by which means these restrictions are counteracted by viruses. In addition to exogenous viruses, we are asking how endogenous retroviruses and mobile genetic elements are recognized and controlled by intrinsic immune factors and how these interactions prevent autoimmunity, inflammation, and tumorigenesis.
The antiretroviral restriction factor SAMHD1
The SAM and HD domain-containing protein 1 (SAMHD1) is a dNTP triphosphohydrolase that plays a crucial role in a variety of different cellular functions. Besides balancing intracellular dNTP concentrations, facilitating DNA damage repair, and dampening excessive immune responses, SAMHD1 has been shown to act as a major restriction factor against various virus species. In addition to its well-described activity against retroviruses such as HIV-1, SAMHD1 has been identified to also reduce the infectivity of different DNA viruses such as the herpesviruses CMV and EBV, the poxvirus VACV, or the hepadnavirus HBV. While some viruses are efficiently restricted by SAMHD1, others have developed evasion mechanisms that antagonize the anti-viral activity of SAMHD1.
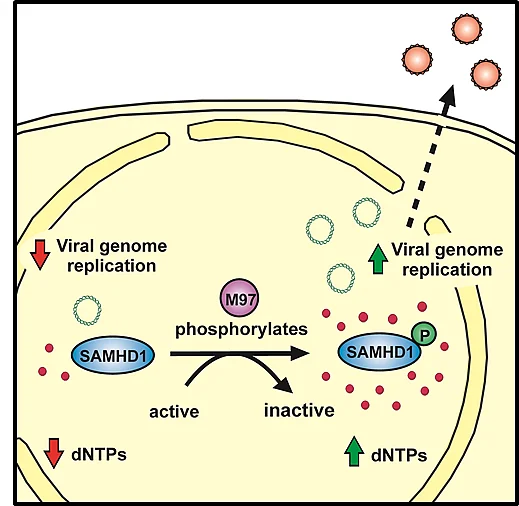
The viral kinase M97 counteracts the in vivo restriction of murine cytomegalovirus by SAMHD1
Human cytomegalovirus belongs to the herpesvirus family and is of great clinical importance, especially during pregnancy or in immunosuppressed patients such as transplant recipients. Although the antiviral activity of SAMHD1 towards DNA viruses has been demonstrated, the role of SAMHD1 during cytomegalovirus (CMV) infection remains elusive. To determine the impact of SAMHD1 on CMV replication, we used murine CMV (MCMV) to infect our previously established SAMHD1 KO mouse model and found that SAMHD1 inhibits MCMV replication in vivo. We report that murine CMV has evolved a tool to counteract SAMHD1 and employs a novel mechanism to inactivate the restriction factor. By comparing MCMV replication in vitro in myeloid cells and fibroblasts from SAMHD1 KO and control mice, we found that the viral kinase, M97, actively counteracts SAMHD1 by inducing its phosphorylation at the regulatory residue threonine 603, thereby inactivating SAMHD1. The phosphorylation of SAMHD1 in infected cells correlated with a reduced dNTP hydrolase activity and the loss of viral restriction. In addition, we showed for the first time that SAMHD1 acts as a bona fide restriction factor against a naturally occurring virus in vivo. Using our previously established SAMHD1 KO mouse model, we found that SAMHD1 blocks the in vitro and in vivo replication of murine CMV, the well-established model for the human pathogen HCMV. Together, we demonstrate that SAMHD1 acts as a restriction factor in vivo and identify the M97-mediated phosphorylation of SAMHD1 as a so far undescribed viral countermeasure. Our findings explain how CMV is able to replicate in its target cells and demonstrate the importance of the inactivation of SAMHD1 by the viral kinase for in vivo replication, which makes the M97-mediated inactivation of SAMHD1 an attractive drug target.
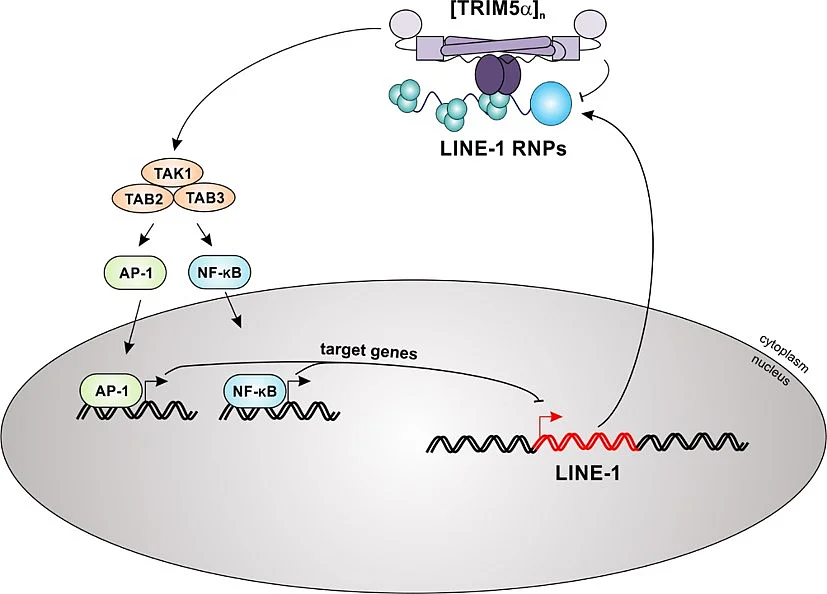
TRIM5α colocalizes and interacts with LINE-1 RNPs in the cytoplasm via its SPRY domain. The sensing of LINE-1 RNPs initiates the multimerization of TRIM5α and the downstream activation of the TAK1 complex. Subsequently, activated MAP kinase and NF-κB signaling cascades lead to a block of LINE-1 retrotransposition, most likely via expression of AP-1 and NF-κB target genes that down-regulate LINE-1 promoter activity.
TRIM5α blocks and senses LINE-1 retroelements
We found that the host factor TRIM5α senses and restricts LINE-1 retroelements. Previously, TRIM5α has mainly been described as antiretroviral restriction factor. Most notably, rhesus TRIM5α is well-known to efficiently block HIV-1 replication, while human TRIM5α is less active. Surprisingly, we found that both human and rhesus TRIM5α efficiently repress human LINE-1 retrotransposition. TRIM5α interacts with LINE-1 ribonucleoprotein complexes in the cytoplasm, which is essential for restriction. In addition, we found that TRIM5α induces innate immune signaling (AP-1; NF-κB) upon interaction with LINE-1. Thus, our findings identify TRIM5α as pattern recognition receptor and restriction factor against endogenous mobile genetic elements, such as LINE-1. Recently, we were able to confirm and extend this activity. We found that a frequent polymorphism (SNP) in TRIM5α, H43Y, blocks LINE-1 retrotransposition with even higher efficiency than WT TRIM5α. Upon sensing of LINE-1 complexes in the cytoplasm, TRIM5α H43Y activates both NF-kB and AP-1 signaling pathways more potently than the WT allele, thereby triggering a strong block to the LINE-1 promoter. Interestingly, the H43Y allele lost its antiviral function suggesting that its enhanced activity against endogenous LINE-1 elements is the driving force behind its maintenance within the population. Thus, our study suggests that the H43Y variant of the restriction factor and sensor TRIM5α persists within the human population since it preserves our genome from uncontrolled LINE-1 retrotransposition with higher efficiency.
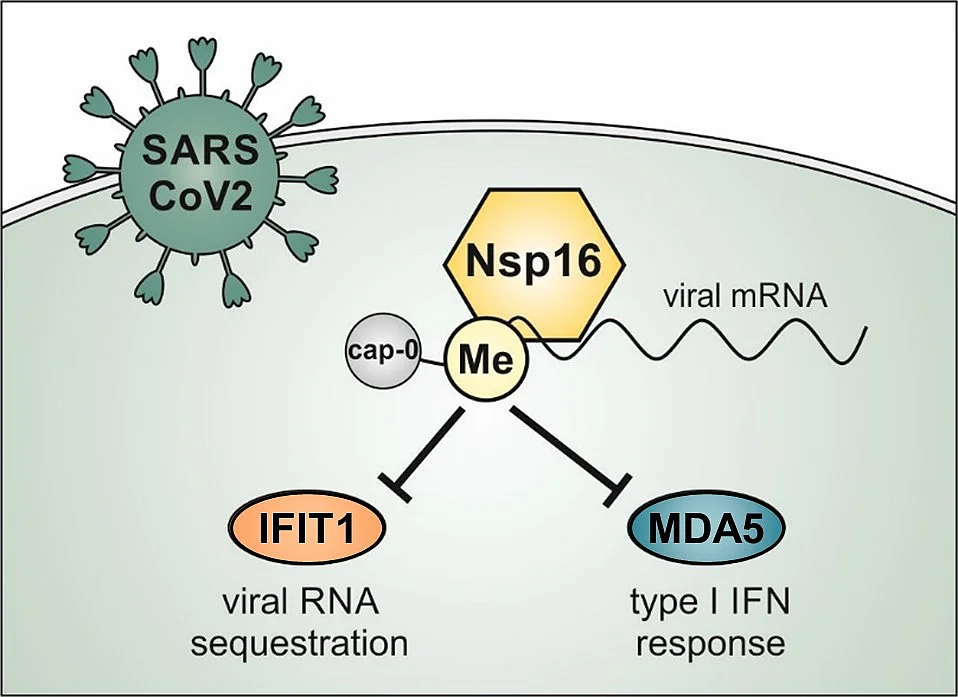
Nsp16 shields SARS–CoV-2 from efficient MDA5 sensing and IFIT1-mediated restriction
Methylation of the mRNA cap by cellular methyltransferases enables efficient translation and avoids recognition by innate immune factors. Coronaviruses encode viral 2’-O-methyltransferases to shield their RNA from host factors. Recently, we generated recombinant SARS–CoV-2 harboring a catalytically inactive 2’-O-methyltransferase Nsp16, Nsp16mut, and analyzed viral replication in human lung epithelial cells. We found SARS–CoV-2 Nsp16mut to be highly immunogenic, resulting in a strongly enhanced release of type I interferon upon infection. The elevated immunogenicity of Nsp16mut is absent in cells lacking the RNA sensor MDA5. Also, we found that Nsp16mut is highly sensitive to type I IFN treatment and demonstrated that this strong antiviral effect of type I IFN is mediated by the restriction factor IFIT1. Together, we described a dual role for the 2’-O-methyltransferase Nsp16 during SARS-CoV-2 replication. It helps to avoid efficient recognition by MDA5 and shields viral RNA from interferon-induced antiviral responses, Together, we identified Nsp16 as a promising target for generating attenuated and highly immunogenic SARS-CoV-2 strains and as a potential candidate for therapeutic intervention.

