
FAU-Nachwuchsgruppe Retroviral Pathogenesis: HTLV-1 and Adult T-Cell Leukemia
More details: https://www.thoma-kress-lab.de/
Human T-cell leukemia virus Type 1 (HTLV-1)
Our group studies HTLV-1, a highly oncogenic yet neglected retrovirus, which primarily infects white blood cells (CD4+ T-cells) in vivo and causes incurable diseases like Adult T-cell leukemia/ lymphoma (ATLL) or inflammatory maladies like HTLV-1-associated myelopathy/ tropical spastic paraparesis (HAM/TSP) after lifelong viral persistence. Worldwide, at least 5-10 million people are HTLV-1 infected and most of them are unaware of their infection. Endemic regions are located in Japan, Central Australia, Melanesia, South America, the Caribbean, and the Middle East. The virus is transmitted via cell-containing body fluids such as blood products, semen and breast-milk, which constitutes the major route of mother-to-child transmission. After infection of CD4+ T-cells, HTLV-1 is reversely transcribed and integrates into the host cell genome. Despite the high number of people worldwide living with a persistent HTLV-1 infection, there is still no efficient prevention strategy, vaccine or cure for HTLV-1 and its associated diseases.
Our major research aims are:
- Understand breast milk transmission of HTLV-1 and develop prevention strategies
- Elucidate molecular mechanisms of HTLV-1 cell-to-cell transmission
- Develop strategies to lower the proviral load of infected individuals
- Interfere with HTLV-1 induced leukemogenesis
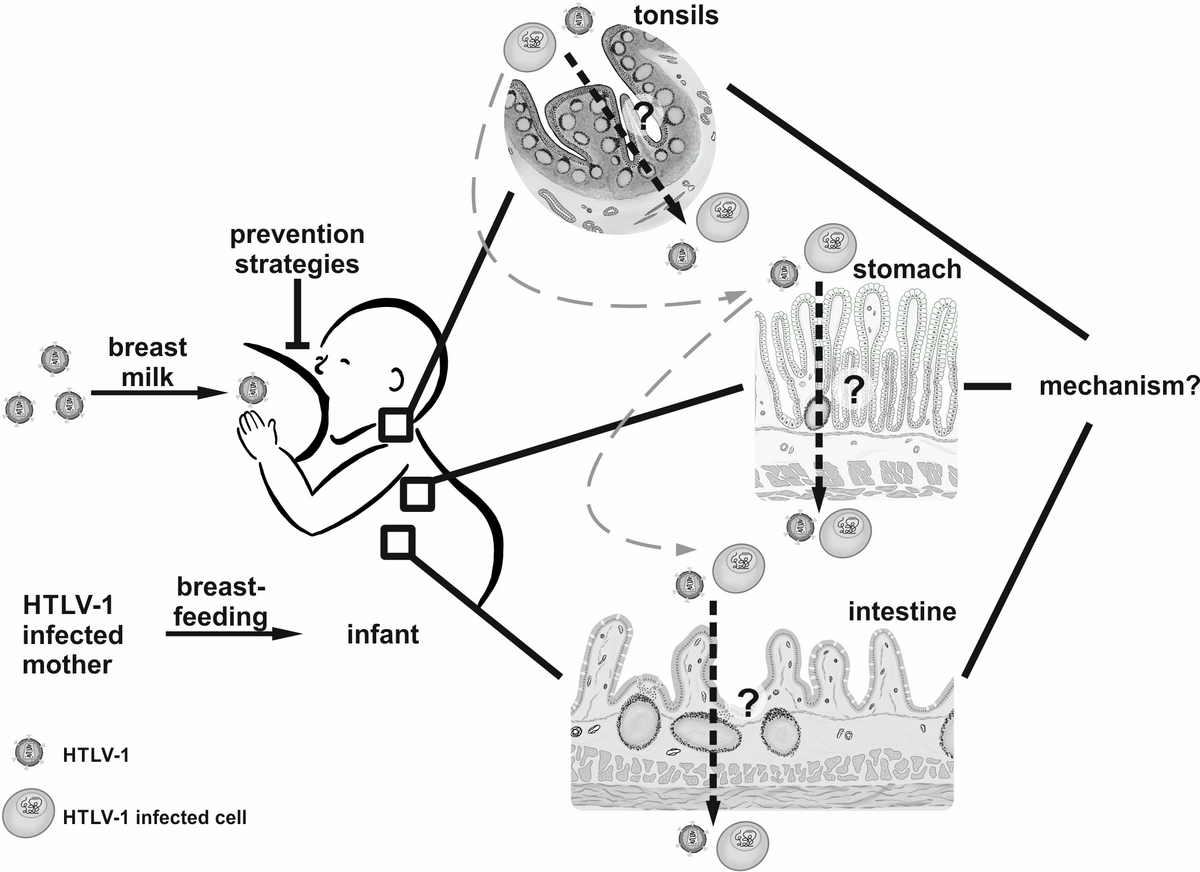
Milk transmission of HTLV-1 and develop prevention strategies
At the end of 2020, we started a new area of research in our group since the Ministry of Education and Research (BMBF) has supported us by funding a Junior Research Group in Infection Research. Within the project Milk-TV, which is short for “Milk Transmission of Viruses”, we seek to explore early events of HTLV-1 transmission and to develop prevention strategies to fight HTLV-1 infections. Briefly, breastfeeding is recommended by the World Health Organization for at least 6 months and up to 2 years of age, and it is proven that breast milk protects against several diseases and viral infections. Intriguingly, few viruses are preferentially transmitted via breastfeeding including HTLV-1. Risk of transmission increases with the duration of breastfeeding, however, abstinence from breastfeeding is not an option in resource-limited settings or underrepresented areas or populations. Despite significant progress in understanding details of cell-to-cell transmission, it is still unclear, which cells in which organs get infected via the oral route of virus transmission (Kemeter et al., 2023), how these cells get infected, how breast milk affects this route of infection, and how to inhibit oral transmission despite breastfeeding, which is an urgent need, especially in underrepresented areas of the world (Figure 1, Millen and Thoma-Kress, 2022). Thus, we strive within this project to address these questions experimentally with the help of different tissue culture model systems and the development of strategies to block HTLV-1 transmission. Overall, our central aim is to uncover prevention strategies that might ultimately allow infants to benefit from breastfeeding while reducing the risk of HTLV-1 transmission.
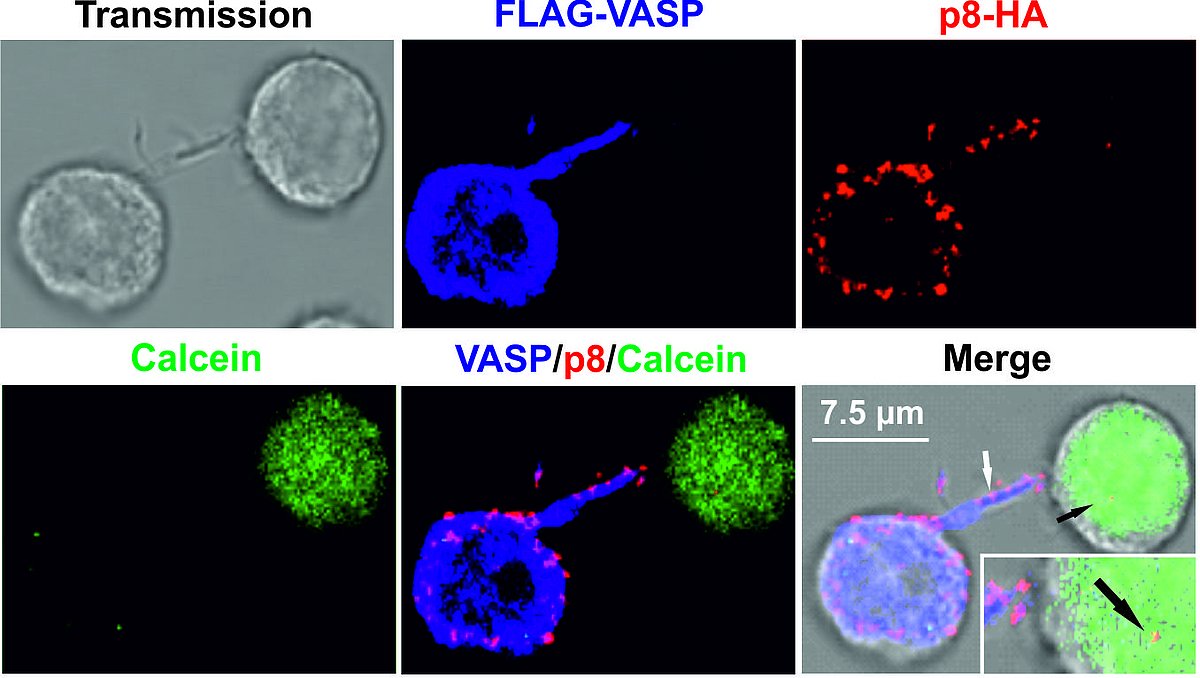
Elucidate molecular mechanisms of HTLV-1 cell-to-cell transmission
Transmission of HTLV-1 to other T-cells is strictly dependent on cell-cell contacts, and viral particles are transferred after polarized budding at a tight and confined cell-cell contact, the so-called virological synapse. Earlier work of our group identified the actin-bundling protein Fascin to be crucial for HTLV-1 cell-to-cell transmission (Gross et al., 2016). During the last years, we studied transcriptional regulation of Fascin in more detail. Interestingly, HTLV-1 and related tumor viruses have evolved strategies to transcriptionally induce Fascin (Heym et al., 2022). A fragment of the human Fascin promoter, which we identified to specifically respond to HTLV-1 (Mohr et al., 2015), was also suitable for transcriptional targeting of dendritic cells, which might be useful in vaccination approaches (Zeyn et al., 2023).
HTLV-1 transmission may also occur via viral biofilms at virological synapses, an agglomeration of viruses that are packaged on top of the infected cells in a microbial biofilm-like structure. Our work shed further light on the composition of theses viral biofilms. Based on transcriptomics and data mining, we found that the structural protein Collagen IV (COL4A1/ COL4A2) is transcriptionally regulated by Tax and a component of viral biofilms, with COL4A2 appearing to impact virus transmission. Thus, our study provided first links between Tax’s activity and formation of the viral biofilm by hijacking COL4 protein functions (Millen et al., 2019).
HTLV-1 is not only transmitted at tight cell-cell contacts, rather, transmission may also occur via long-distance connections, which are induced by the small accessory viral protein p8. This protein is generated by proteolytic cleavage from the precursor p12 encoded on open reading frame I of the HTLV-1 genome. p8 induces cellular protrusions and is transferred to other cells to foster HTLV-1 cell-to-cell transmission. To get molecular insights into p8 transfer and HTLV-1 transmission, we made use of bioinformatics in combination with proteomics. Our studies identified novel interaction partners of p8 (Figure 2), amongst them, vasodilator stimulated phosphoprotein (VASP). After having developed a novel flow cytometry-based assay which allows rapid and automatic quantitation of p8 transfer to target cells (Donhauser et al., 2018), we could show that knockout of VASP delays the transfer of p8 between cells, most likely by impairing recruitment of p8 to the plasma membrane (Donhauser et al., 2020). Importantly, this study revealed that VASP is also important for HTLV-1 cell-to-cell-transmission.
Develop strategies to lower the proviral load of infected individuals
Our lab works on different projects to interfere with HTLV-1 persistence to ultimately lower the proviral load. This is of special importance since the risk to develop HTLV-1 associated diseases increases with higher proviral loads. Upon infection with HTLV-1, the viral transactivator Tax stimulates viral replication by recruiting host cell factors like positive transcription elongation factor b (p-TEFb) to the viral promoter and by enhancing mitotic expansion of infected cells. Since viral gene expression is repressed in vivo by viral, cellular, and epigenetic mechanisms in late phases of infection, HTLV-1 avoids an efficient CD8+ cytotoxic T-cell (CTL) response directed against the immunodominant viral Tax antigen. Hence, new therapeutic strategies aim to transiently activate viral gene expression to shift the equilibrium in favor of an enhanced CTL response to enhance immunogenicity of HTLV-1 Tax, and thus, to expose the latent HTLV-1 reservoir to immune destruction. Thus far, the composition of the protein complex guiding viral gene expression is only partially understood, and systematic analyses and comparisons of compounds affecting viral transcription are lacking. In earlier work, we could identify strong and specific upregulation of the transcription elongation factor ELL2 in HTLV-1-infected cells (Mann et al., 2014). We found that ELL2 strongly enhances Tax-mediated transactivation of the HTLV-1 promoter, and that ELL2 and Tax are part of a common protein complex. However, the detailed composition of this complex and its impact on viral reactivation were unclear. We recently uncovered important domains within Tax and ELL2 which are crucial for Tax:ELL2 complex formation (Kohrt et al., 2021). Moreover, we aim to develop strategies to interfere with HTLV-1 transcription, and, thus, to enhance immunogenicity of HTLV-1 (Schnell et al., 2021). Among several selected histone deacetylase inhibitors, we identified Panobinostat and Romidepsin to be superior to previously tested compounds in enhancing viral transcription (Schnell et al., 2022), however, their impact on viral protein expression was only moderate. Thus, we worked on alternative strategies to interfere with HTLV-1. In a collaborative project, we attacked the integrated HTLV-1 provirus by molecular evolution of a designer recombinase, which was adapted to a recognition site within the HTLV-1 long terminal repeats flanking the integrated proviral DNA (Rojo-Romanos et al., 2023).
Interfere with HTLV-1-induced leukemogenesis
It has been known for years that constitutive activation of the classical and alternative NF-κB signaling pathways by the viral oncoprotein Tax is a hallmark of HTLV-1-driven cancer. NF-κB-deficient Tax transgenic mice lack the induction of ATL-associated aggressive skin diseases. Further, animal studies therapeutically targeting NF-κB slow down and reduce tumor growth in ATL-like diseases. Although there are different reports whether NF-κB is critical for initiating cellular transformation, there is a strong connection between Tax, NF-κB, tumor formation and maintenance. We could recently show that activation of NF-κB signaling specifically enhances the abundance of Tax protein, but not of Tax transcripts (Millen et al., 2020). This led to the identification of a positive feedback loop between Tax and NF-κB activity, which results in enhanced protein expression of Tax and might thus serve as a novel therapeutic target to interfere with Tax-driven transformation. In a collaboration with Dr. Calabro (Padova, Italy), we seek to elucidate the interplay between HTLV-1-infected T-cells and the tumor microenvironment making use of a mouse model of Adult T-cell leukemia.
AG Thoma-Kress in the media:
Meilenstein im Kampf gegen Auslöser einer Form der Leukämie. 28.03.2023.
https://www.virologie.uk-erlangen.de/aktuelles/nachrichten/detail/meilenstein-im-kampf-gegen-ausloeser-einer-form-der-leukaemie/
Neue FAU-Nachwuchsgruppenleiterin: Dr. Andrea Thoma-Kreß. 30.3.2021.
https://blogs.fau.de/gsfau/2021/03/neue-nachwuchsgruppenleiterin-dr-andrea-thoma-kress/
Unsere FAU-Nachwuchsgruppenleitungen. 30.3.2021
Unsere FAU-Nachwuchsgruppenleitungen | Friedrich-Alexander-Universität Erlangen-Nürnberg
Wege in die Wissenschaft. 24.2.2021. FAU-Magazin “Friedrich”.
https://www.fau.de/2021/02/friedrich/wege-in-die-wissenschaft/
Virus im Fokus. Infektionsforschungsprojekt profitiert von Nachwuchsgruppenförderung durch das BMBF. 16.11.2021.
https://www.fau.de/2020/11/news/wissenschaft/virus-im-fokus/
Der Weg ist das Ziel. Ampuls 3/2020, Seite 7. Zeitung für alle Mitarbeitenden des Uni-Klinikums Erlangen.
https://mitarbeiterportal.intranet.uk-erlangen.de/display/AKTUELLESpub/Archiv?preview=/5715115/158270003/ampuls_2020_3.pdf
Auszeichnung für herausragende Grundlagenforschung zur Tumorentstehung. 5.10.2017.
https://www.fau.de/2017/10/news/panorama/auszeichnung-fuer-herausragende-grundlagenforschung-zur-tumorentstehung/
Twitter: @ThomaKressLab
https://twitter.com/ThomaKressLab

