
Protein Kinases as Regulators of Herpesvirus Infections
The human cytomegalovirus (HCMV) is a major opportunistic pathogen of humans with a worldwide distribution. Primary infection with HCMV in persons with a normal immune system is generally mild or asymptomatic. However, HCMV frequently causes systemic disease with severe clinical consequences in immunocompromised persons and in the situation of congenital infection during pregnancy (cCMV). Most of the currently available anti-HCMV drugs face limitations in terms of unwarranted side-effects and the induction of drug resistance. Thus, the development of novel anti-HCMV drugs, and similarly also broader-acting antiherpesviral drugs, is urgently required. For this, protein kinase inhibitors proved to be highly interesting drug candidates, as both cellular and herpesviral protein kinases represent important determinants of viral replication and pathogenicity.
Research objectives
The research concept of the group is focused on the following topics:
- Functional and structural characterization of the HCMV protein kinase pUL97
- Cross-talk between herpesviral and cellular kinases
- Nuclear egress of HCMV and other herpesviruses
- Cyclin–kinase complexes modulated by viral infection
- Virus–host protein assemblies as factors of viral pathogenesis
- Drugs, targets/kinases, and mechanisms: new antiviral strategies.
Functional and structural analysis of the cytomegalovirus-encoded protein kinase pUL97
In general, herpesviral protein kinases are important determinants of the efficiency of viral replication. They fulfill a number of regulatory functions by phosphorylating both viral and cellular proteins. Specifically, the pUL97 kinase of HCMV is the prototype of the UL-subfamily of herpesviral serine/threonine-specific protein kinases (distinct from the HSV-1 US-subfamily). An analysis of the structure–activity relationship of pUL97 and homologous kinases of other herpesviruses resulted in the definition of the kinase domain, the mapping of functionally important regions and the identification of phosphorylated substrates. Experimental settings to reveal the 3D structure of pUL97 are presently performed, including both approaches of structural biology and bioinformatics. As an important functional feature of pUL97, we demonstrated that this kinase is expressed in three isoforms arising from alternative translational initiation. Catalytic activity, including autophosphorylation and substrate phosphorylation, proved to be indistinguishable for the first two isoforms (M1 and M74), while the third isoform (M157) comprised functional differences. In particular, the interaction and phosphorylation of viral substrate proteins was less efficiently performed by the smallest isoform M157. These findings were substantiated by the analysis of recombinant HCMVs expressing individual pUL97 isoforms.
The term 'vCDK': herpesviral orthologs of host cyclin-dependent kinases
A very relevant consequence of the investigations of herpesviral kinases was the recognition of similarities with cyclin-dependent kinases (CDKs) of the host cell. As mainly based on findings reported for the HCMV-encoded protein kinase, four lines of evidence created the new term of vCDK/pUL97. First, a report of our group described the structural similarity between pUL97 and CDKs. In the absence of any crystallization-based structural information about herpesviral kinases, the bioinformatic-driven structural modeling revealed a number of comparable features, when using known 3D structures of host CDKs as modeling templates. Second, the use of a CDK-depleted yeast mutant revealed a partial functional complementation by heterologous overexpression of the viral pUL97. Third, the identification of phosphorylated substrates pUL97 more and more revealed that these contain typical CDK substrates, such as retinoblastoma protein, nuclear lamins A/C, and others. Finally, our closer investigations of host protein interaction of HCMV pUL97 provided evidence that human cyclins, in particular types H, T1 and B1, represent specific binding partners. Taken together, these properties pointed to the classification of pUL97 and related herpesviral kinases as vCDKs.
For details see Tillmanns, Kicuntod et al. (2024), Int. J. Mol. Sci.
The regulatory importance of protein kinases for cytomegalovirus nuclear egress
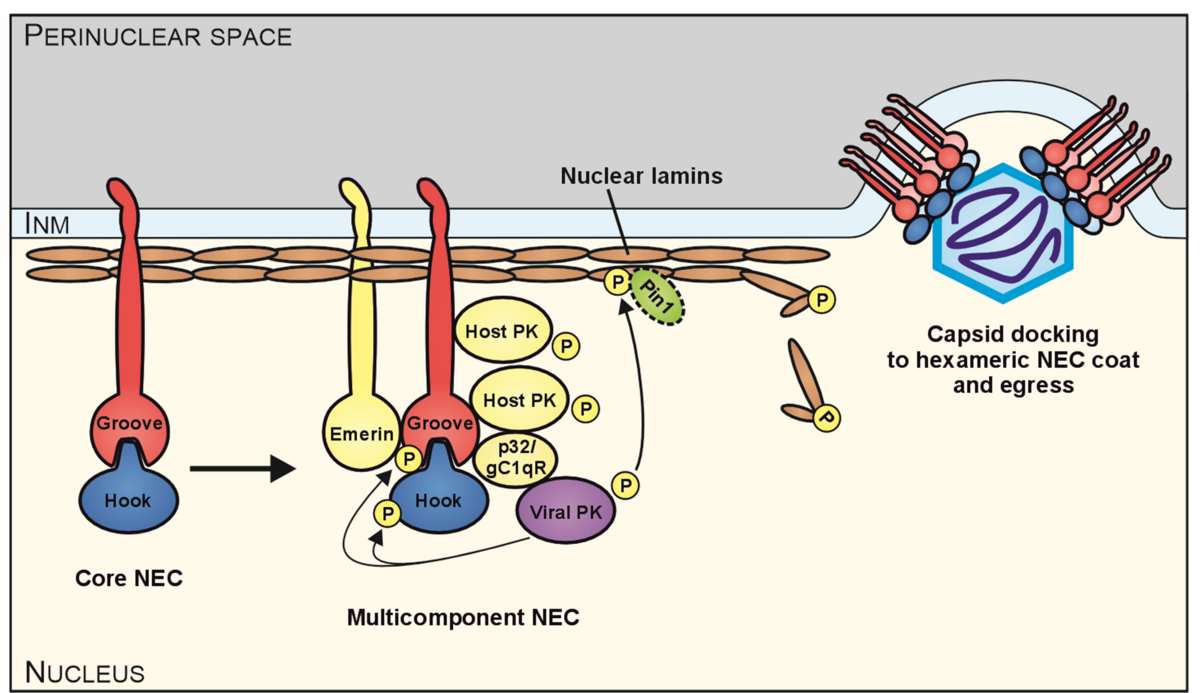
The role of pUL97 within the HCMV replication cycle is manifested in several different stages, including viral DNA synthesis, phosphorylation-mediated inactivation of the cell cycle regulator Rb and nuclear viral capsid egress. Concerning the nuclear capsid egress, we demonstrated an involvement of pUL97 in the phosphorylation of nuclear lamins. The nuclear lamina is a rigid proteinaceous meshwork underlining the inner nuclear membrane. During herpesviral infections, the nuclear lamina restricts the efficiency of the nucleocytoplasmic transport of viral capsids, because of the large size of herpesviral capsids, which does not allow a direct transition through the nuclear pore complex. Lamina destabilization requires site-specific phosphorylation of lamins and lamin-binding membrane proteins. In case of HCMV, we demonstrated that pUL97 is recruited to the nuclear lamina through the interaction with a cellular multi-ligand binding protein, p32/gC1qR, which is also associated with the nuclear transmembrane protein emerin. pUL97 subsequently phosphorylates lamins of type A/C in a site-specific manner (Ser22) to induce a morphological alteration of the nuclear lamina. Thus, pUL97 has a crucial effect on the destabilization of the lamina, which is a prerequisite for the viral nuclear egress. In addition to pUL97, two other viral proteins are strictly associated with the nuclear lamina, namely pUL50 (groove protein) and pUL53 (hook protein). The pUL50–pUL53 interactor pair is considered as core of the HCMV-specific nuclear egress complex (NEC) that recruits a number of additional viral and cellular proteins towards a multicomponent complex. Importantly, the Ser22-specific lamin phosphorylation generates a binding motif for the cellular prolin-specific isomerase Pin1 that induces a cis/trans isomerization of Pro23 in lamin A/C. According to the present state of the art, the Pin1 activity represents the main mechanism of the herpesvirus-induced structural conversion of the nuclear lamina, thus leading to local lamina disassembly as a prerequisite for viral nuclear capsid egress (Figure 1).
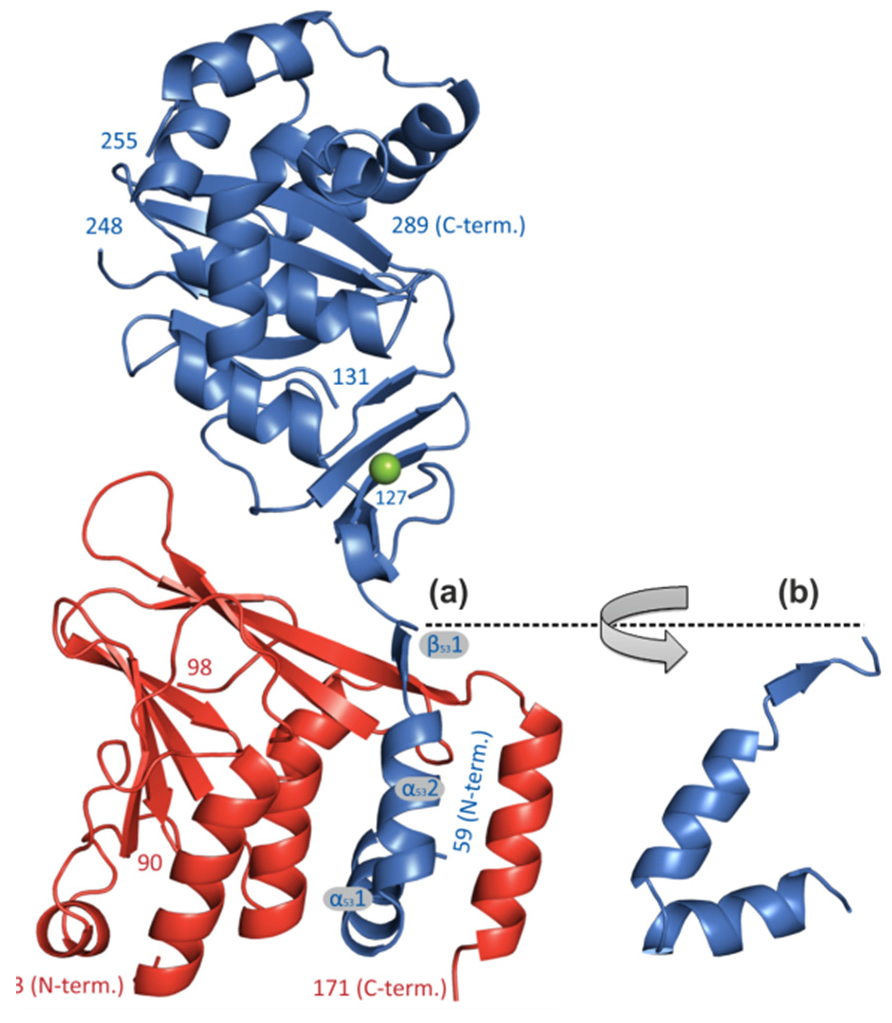
The secondary structure elements of the hook-like N-terminal extension of pUL53 are presented in two different orientations (a) and (b), thus embracing pUL50 and becoming buried in the interface formed by three pUL50 -helices (ribbon representation: pUL50, red; pUL53, blue). For details see Walzer et al., 2015, JBC.
Moreover, we were able to characterize the pUL50–pUL53 nuclear import and heterodimerization at the inner nuclear membrane. While pUL50 is a trans-membrane protein, directly inserted into the inner nuclear membrane, pUL53 is translocated to the nucleus via its recently mapped N-terminal NLS and is recruited to the inner nuclear rim by binding to pUL50. Thus, direct pUL50–pUL53 interaction is a prerequisite for intracellular trafficking and the fine-localization of both proteins. A hallmark of these investigations was the successful crystallization and resolution of the 3D structure of the globular domains of the pUL50–pUL53 core NEC of HCMV and other herpesviruses by our group and other investigators. The pUL53-specific features include a zinc-binding site and a hook-like N-terminal extension, the latter representing a crucial element of the pUL50–pUL53 interaction. The hook-like extension (amino acids 59–87) embraces pUL50 and contributes 1510 Å2 to the total interface area (hook-into-groove interaction; Figure 2). The pUL50 structure reveals a considerable repositioning of its aC helix upon pUL53 binding. The resolution of additional herpesviral NECs, namely EBV (Muller et al., 2020, JBC) and VZV (Schweininger et al., 2022, JBC) by our group confirmed this unique NEC property, termed as herpesviral hook-into-groove interaction. A close examination of the crystal structure indicated partial assembly of pUL50–pUL53 heterodimers to hexameric ring-like structures possibly providing additional scaffolding opportunities for the multimeric NEC. Moreover, our biochemical and proteomic investigations of binding partners of the pUL50–pUL53 complex suggested a combination of viral and cellular constituents associated with the multimeric NEC in its final composition at late stages of HCMV replication. In particular for pUL50, five binding partners were identified, i.e. pUL53, PKCa, p32/gC1qR, emerin and CDK1. A phospho-specific mass spectrometry analysis led to the identification of 14 major and minor sites of pUL50 phosphorylation (as putative sites for the pUL50-phosphorylating kinases pUL97, PKCa and CDK1).
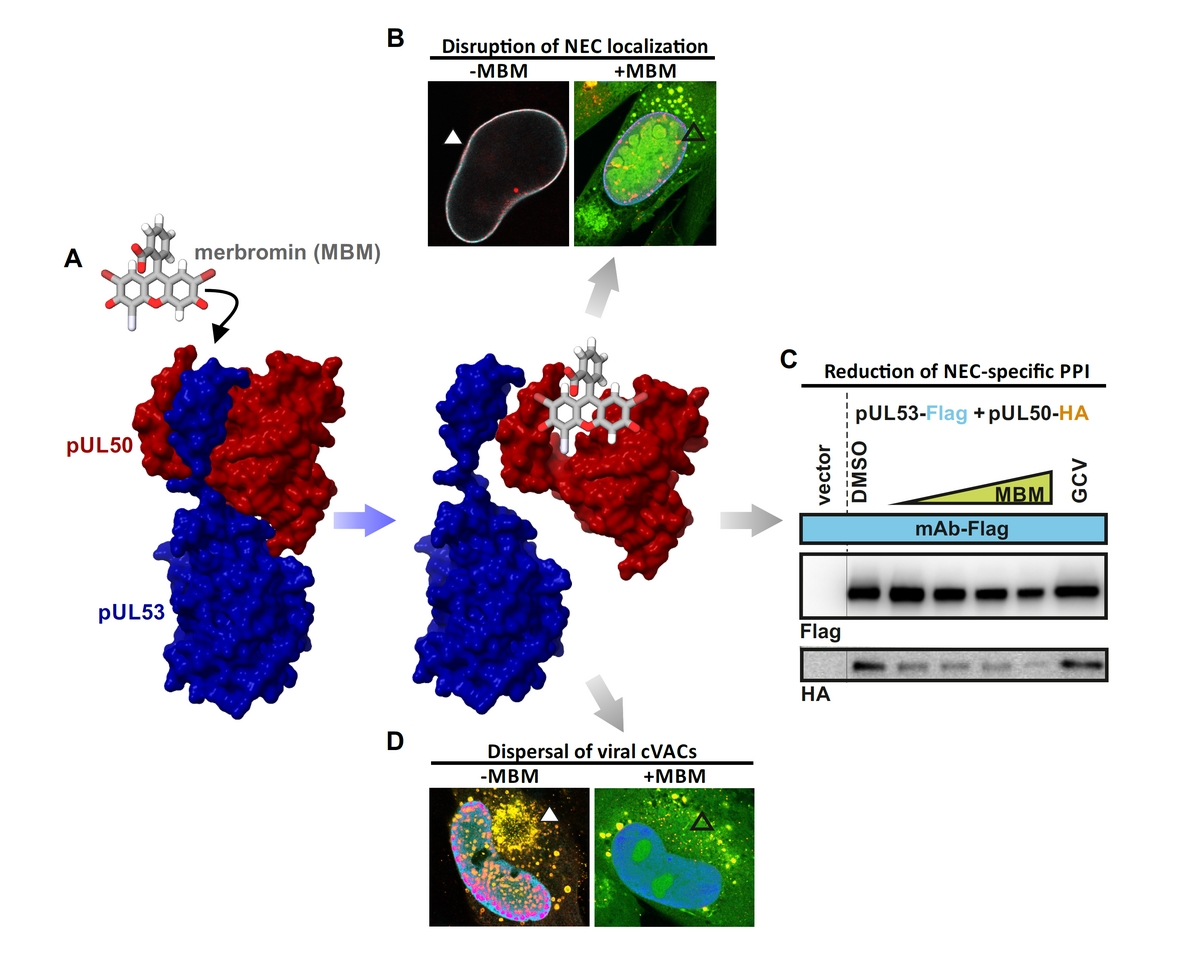
(A) Chemical structure of merbromin (MBM). (B) MBM disrupts the localization of pUL53 on the nuclear rim. (C) The interaction of HCMV core NEC proteins, pUL50–pUL53, decreases in the presence of higher concentrations of MBM. (D) The localization of the cytoplasmic viral assembly complex (cVAC) is dispersed after treatment with MBM.
The ongoing functional investigation of the HCMV-specific NEC may reveal novel insights into these pronounced functional ties of virus-host interaction. As far as current options of NEC-specific antiviral targeting are concerned, we see a number of highly relevant aspects: (i) the blocking of core NEC formation, (ii) the interference with activities of the multicomponent NEC (such as site-specific phosphorylation or inhibition of lamin-directed prolyl cis/trans isomerization), (iii) the interference with capsid-docking to the NEC, or (iv) the blocking of membrane specific activities during primary envelopment. Regarding the situation of a multifactorial regulation of the herpesviral nuclear egress-specific mechanism, additional strategies of NEC interference appear possible. It should be emphasized that we already identified the first prototypes of NEC-inhibitory small molecules, such as merbromin (MBM; Figure 3), ibrutinib (IBM), LDC599, and others. Along with the characterization of these new core NEC inhibitors, we recognized that the property of covalent target binding of compounds proved to be highly advantageous for the efficacy of PPI inhibition. Therefore, the study of these covalent binders, termed 'warheads', recently attracted our closer interest. The mechanistic analyses of warheads, either directed against NEC, kinases or other regulatory viral proteins are presently ongoing. Moreover, an indirect NEC-associated prototype inhibitor was characterized with maribavir (MBV). This drug is directed to the viral kinase pUL97, which is a crucial codeterminant of NEC regulation, so that MBV exerts a strong nuclear egress-inhibitory activity. MBV has recently been clinically approved as the only kinase inhibitor within the entire field of antiviral therapy.
Cyclins and cyclin-dependent protein kinases (CDKs) serve as coregulators of cytomegalovirus replication
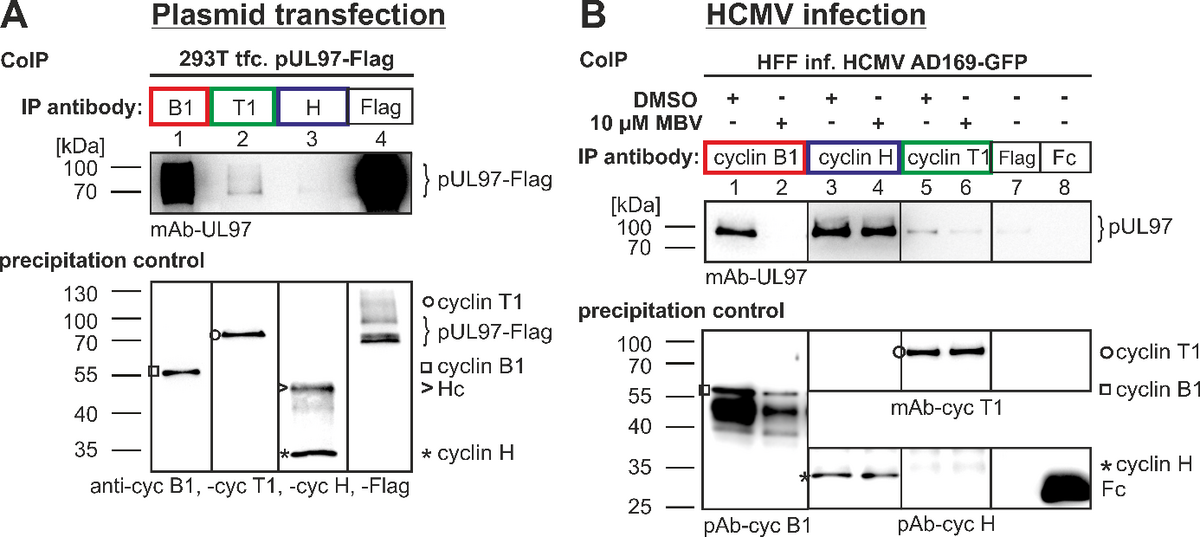
An important focus of our investigations is the identification of central phosphorylation pathways of cellular protein kinases which are essential for the replication of herpesviruses. In this regard, the phosphorylation of viral regulator proteins by cellular protein kinases is particularly interesting. In the case of HCMV, the specific interaction between viral proteins and cellular protein kinases, especially cyclin-dependent protein kinases (CDKs), is presently investigated. The identification of regulatory consequences of such interactions will define novel aspects of virus-host cell interaction. Our recent data confirmed previous reports that CDK activity modulates the intracellular localization of the HCMV nuclear regulatory protein pUL69. CDK inhibitors induce a nuclear speckled aggregation of pUL69 in HCMV-infected fibroblasts. Moreover, experimental data proved a direct protein interaction between pUL69 and CDK9/cyclin T. Interestingly, we identified that the phosphorylation of pUL69 can be mediated through cellular CDK as well as viral pUL97 kinase activities. This finding supported the current postulate that pUL97 represents a viral CDK ortholog. We identified additional interactions between pUL97 and human cyclins of types T1, B1 and H (Figure 4).
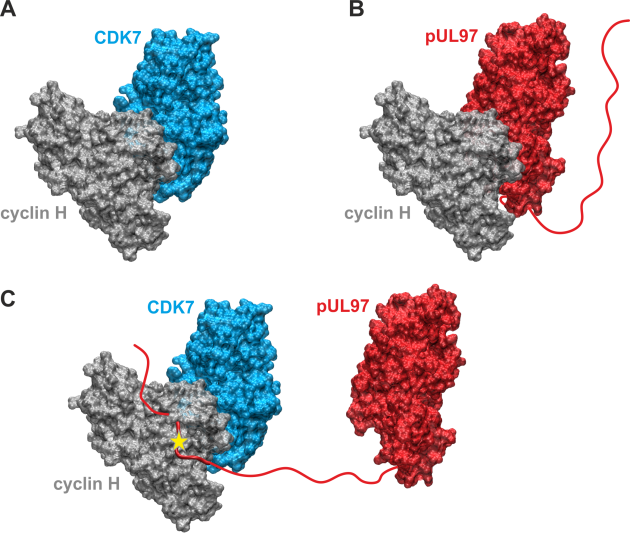
(A) Model of the binary CDK7-cyclin H complex. (B) Model of the binary pUL97-cyclin H complex. Both models were generated based on the homologous CDK9-cyclin T1 complex crystal structure using the strategy described before and revealed the canonical interaction via the large globular domain interfaces. The globular domain interfaces shown in (A) and (B) are overlapping and thus only one partner can interact with cyclin H via this interface. (C) Model of a putative ternary CDK7-cyclin H-pUL97 complex, in which the long unstructured N-terminal region of pUL97 is supposed to interact with cyclin H via an alternative binding motif or interface. A yellow star denotes a yet undefined modification of cyclin H induced by the HCMV-specific infectious environment that is required for pUL97 binding.
As a specifically important aspect, the demonstration of pUL97-cyclin complexes by biochemical settings (CoIP), mass spectrometry-based proteomics and bioinformatic modeling (Figure 5) addressed novel questions of regulatory consequences of the cyclin-associated viral kinase pUL97. Our concept suggests that the cyclins bound to pUL97 may recruit substrate proteins and may reinforce preferential events of pUL97-mediated substrate phosphorylation. Mutational analysis is presently performed and a CoIP-based interactomic analysis of pUL97 site-directed replacement and deletion mutants carrying putative defects in cyclin interaction refined our insight into the underlying regulatory details. Specific findings of the ongoing investigations are: (i) pUL97 interacts with cyclins B1 and H in a manner dependent on pUL97 kinase activity and phosphorylation or HCMV-specific cyclin modulation; (ii) pUL97-mediated in vitro phosphorylation of cyclins is measurable for type B1 but not H, and no evidence is provided for mutual transphosphorylation between pUL97 and CDK7; and (iii) a cyclin T1/H-mediated bridging mechanism of pUL97 self-interaction is supported by current data. Concerning our approaches to define the functional relevance and molecular mode of interaction between pUL97 and human cyclins, we recently identified the following: (i) vCDK/pUL97–cyclin interaction was found conserved in all analyzed HCMV mutants, and viral replication fitness was almost indistinguishable from wildtype; (ii) deletion mutagenesis helped to map the interface 2 (IF2), the deletion of which resulted in a loss of interaction with cyclins T1 and H (but not B1), as well as a severely impaired viral replication efficiency; (iii) upon IF2 deletion, pUL97 kinase activity was significantly reduced, thus proving the functional importance of the pUL97–cyclin interaction; (iv) knock-out and knock-down experiments confirmed the importance of cyclin H as a major determinant of HCMV replication efficiency; (v) a quantitative in vitro kinase assay (qSox-IVKA) showed a significant increase of pUL97 kinase activity proportionally with the presence of cyclin H; and (vi) biochemical and bioinformatic analyses demonstrated a ternary complex vCDK/pUL97–cyclin H–CDK7 (Figure 5), which (vii) enables a transstimulation of CDK7 through pUL97 (but not vice versa).
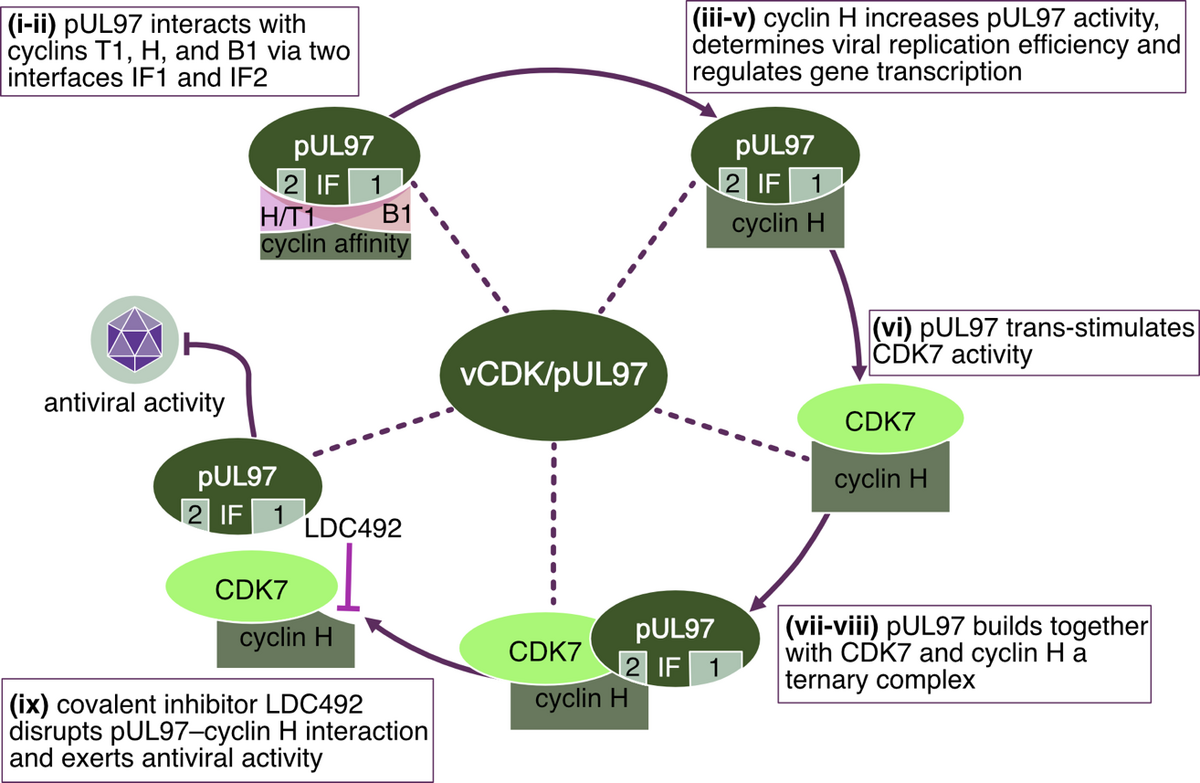
As a consequence of these investigations, a first covalently binding compound that inhibited the pUL97–cyclin H interaction was identified with potent anti-HCMV activity, pointing towards another novel antiviral targeting strategy (Figure 6).
In addition, even more host CDKs have been characterized as coregulators of herpesviral replication by studies of our group and further investigators. Currently, we are performing a characterization of the interaction patterns and regulatory roles of individual CDKs (most of all CDK7) during HCMV infection of human fibroblasts and other cell types. Especially, CDKs and further protein kinases that possess importance for the replication of HCMV as well as other herpesviruses have been considered as potential antiviral drug candidates. In this regard, it appears promising that our current in vitro and also first in vivo data have proven the high antiviral efficacy of inhibitors of virus-relevant CDKs. In particular, a highly potent and selective inhibitor of CDK7, LDC4297, exerts a broad and effective antiviral activity. Interestingly, quantitative approaches of combination treatments identified statistically significant drug synergy levels between inhibitors of CDKs (LDC4297 and others) and vCDK/pUL97 (MBV and others). A mechanistic investigation of the synergy between these drugs pointed to specific advantages of this combination strategy (Figure 7), namely (i) a reduction of effective drug dosages, (ii) a novel kinase-based antiviral mode of action, and (iii) an improvement in drug resistance barriers. According to our current state of investigations, we are describing the further development of enhanced derivatives of parental CDK7 inhibitor, LDC4297, namely QRS compounds with the ability to covalently bind to this host target. Through this modification, an enhancement of anti-HCMV efficacy in vitro has been improved into the picomolar range of drug concentrations. Combined, these findings may be highly beneficial for the development of novel broad-spectrum antiherpesviral drugs.
Virus–host protein interactions: impact on cellular signaling and viral pathogenesis
It is an established concept that cytomegalovirus infection (or similarly infections with many other human herpesviruses) induce a cellular program of signaling that is characterized by the induced expression and activity of a variety of cellular protein kinases. This kind of upregulated signaling program provides a fingerprint, individually reflecting the parameters of replication and pathogenesis of a single virus (virus-specific cellular signaling, VSS). In case of HCMV, we established a novel approach of kinome profiling that suggests an important role of specific cellular protein kinases for viral replication. Experimental inhibition of such kinases showed a significant reduction, whereas inhibition of other kinases led to an activation of HCMV replication. Furthermore, analysis of the mode of action of those kinase inhibitors exerting antiviral activity (VSS inhibitors) suggested a substantial benefit for the efficiency of viral replication at the immediate early or early-late phases of HCMV gene expression. Interestingly, an ongoing comparative analysis of kinase patterns induced by various herpesviruses (as representatives of the alpha-, beta- and gamma-herpesvirus subfamilies) indicated partly conserved events of kinase/signaling upregulation and, for other kinases, virus-specific differences. Thus, our combined data provide new information on host cell kinases involved in viral replication and revealed potential targets for future diagnostic or antiviral proceedings.
The latter aspect is also addressed by our long-term analysis of the antiviral mode of the broad antiinfective drug artesunate. Several experimental findings demonstrated an important point of the antiviral activity of artesunate (ART), i.e. its interference with NF-kB-specific signaling. A direct binding of artesunate to NF-kB p65 is strongly suggested by our data. As an additional mechanistic information arising from our mass spectrometry-based proteomic studies of drug binding (using linker-coupled versions of artesunate compounds), ART and related drugs are able to bind a number of additional regulatory host proteins, such as mitochondrial regulators, a nuclear export factor, and others. Thus, a multifactorial, host-directed mode of antiviral action is strongly supported by these findings. Notably, the enhanced anti-HCMV activity of synthetic artesunate dimers, trimers and hybrid molecules (cooperation with the group of Svetlana B. Tsogoeva) recently opened promising novel perspectives, not only in antiviral research, but also in the mechanistic analysis of HCMV specific cellular signaling. Importantly, the antiviral efficacy of artesunate compounds increased with the degree of multimerization (trimers>dimers>monomers). Also, the functional validation of these drug features and target proteins postulates a regulatory link between mitochondrial activity and the efficiency of HCMV replication.
As another highly attractive point of virus–host protein interaction, we identified the unique role of Pin1 in HCMV replication. Pin1 is a prolyl cis/trans isomerase that converts the conformational cis/trans changes of substrate proteins. Although Pin1 target sites are represented by proline residues (recognized by the Pin1 WW domain), the substrate docking of Pin1 is strongly enhanced through the presence of a phosphorylated serine residue (contained in the pSer/Thr-Pro binding motif). This phospho-dependent mode of Pin1 activity has been demonstrated for the cellular substrate lamin A/C, in which pSer22 determines the rate-limiting step in Pin1-mediated isomerization. The resulting conformational change is a hallmark in the HCMV-specific rearrangement of the host nuclear lamina to facilitate viral nuclear egress. Beyond that, the interaction between Pin1 and viral proteins has also been reported by several researchers including our group. In the case of HCMV, a Pin1-binding activity has been proven for pUL50, pUL69 and pUL44. Hereby, the phospho-specificity of Pin1 binding, as determined by threonine residues T45 and T84 appeared particularly striking. Thus, the regulatory role of Pin1 interaction with pUL44, acting as the processivity factor of viral DNA polymerase, is in the focus of our ongoing research.
Experimental development of antiherpesviral drug candidates based on protein kinase inhibitors
Considering the current repertoire of approved antiherpesviral drugs, it appears striking that the vast majority is provided by direct-acting antivirals (DAAs). This may have practical reasons in the history of antiviral drug development, since the strategy was originally derived from the early recognition of virus-encoded targets and target structures that are unique or differ from their host orthologs. DAAs have been proven to be clinically highly expedient, their achievements and benefits in treatment, however, can be strongly limited. In particular with long-term therapeutic or prophylactic treatments, the induction of viral drug resistance, or unwarranted side-effects. For these reasons, an additional characterization of host-directed antivirals (HDAs) is presently aspired. Host-directed drugs, in particular when recognizing pathogenesis-related mutants of target proteins, have been greatly successful in areas spanning the treatments of tumor, metabolic, infectious, and inflammatory diseases. However, within the field of antiviral therapy, this topic misses in-depth experience, since only quite few HDAs have been clinically approved and utilized in therapeutic settings thus far.
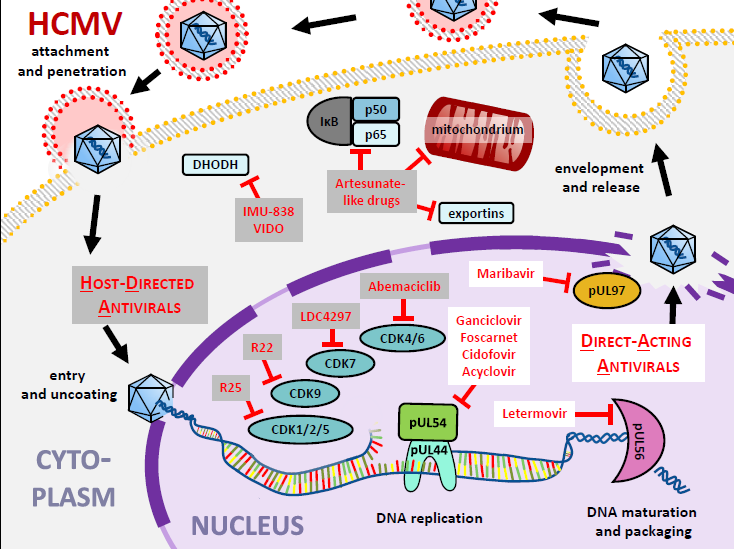
Considering anti-HCMV drug targeting options, both DAAs and HDAs have been analyzed in more detail during the last decades (Figure 8). Hereby the cytomegaloviral kinase pUL97 has been validated as a target enzyme for novel antiviral drugs, with maribavir (MBV) as the first kinase-directed DAA that has recently been approved. This targeting strategy seems particularly interesting due to the fact that pUL97 has already been an essential part of conventional antiviral therapy for a long time, as it carries out a pacemaker phosphorylation reaction on ganciclovir and related nucleoside analogs. For developmental works of the pUL97-directed antiviral strategy in our own group, quantitation systems for pUL97 kinase activity have been established, thereby leading to the identification of novel chemical classes of compounds (quinazolines, indolocarbazoles, benzimi-dazoles, others) that inhibit the pUL97 kinase activity. Hereby, it was shown that these kinase inhibitors block HCMV replication efficiently in vitro and in vivo and that the inhibitory effects on the levels of kinase activity and virus replication are linked. Our study of quinazoline-type inhibitors of pUL97 indicated the high value of bioinformatic-based modeling approaches in the optimization of candidate compounds. An enlarged platform combining biochemistry-, bioinformatic-, structural biology- and medicinal chemistry-based approaches may substantially take forward our efforts of pUL97-directed drug design.
In addition to the analysis of pUL97 inhibitors, a broad-spectrum antiherpesviral activity was also identified for the CDK7 inhibitor LDC4297. This interesting compound comprises an outstanding selectivity for CDK7 (as characterized by a kinome-wide evaluation of >330 kinases profiled in vitro). LDC4297 exerts an efficient inhibition of HCMV replication in primary human fibroblasts at nanomolar concentrations (EC50 24.5±1.3 nM), proved efficacy also in vivo in a MCMV/mouse model, and moreover, provides a broad antiherpesviral activity. In comparison, non-herpesviruses, i.e. adeno-, pox-, retro- and orthomyxoviruses, showed either intermediate or no sensitivity. These results provided a proof-of-concept for the antiviral potency of the targeting of host CDK7 by LDC4297 or even enhanced-efficacy derivatives.
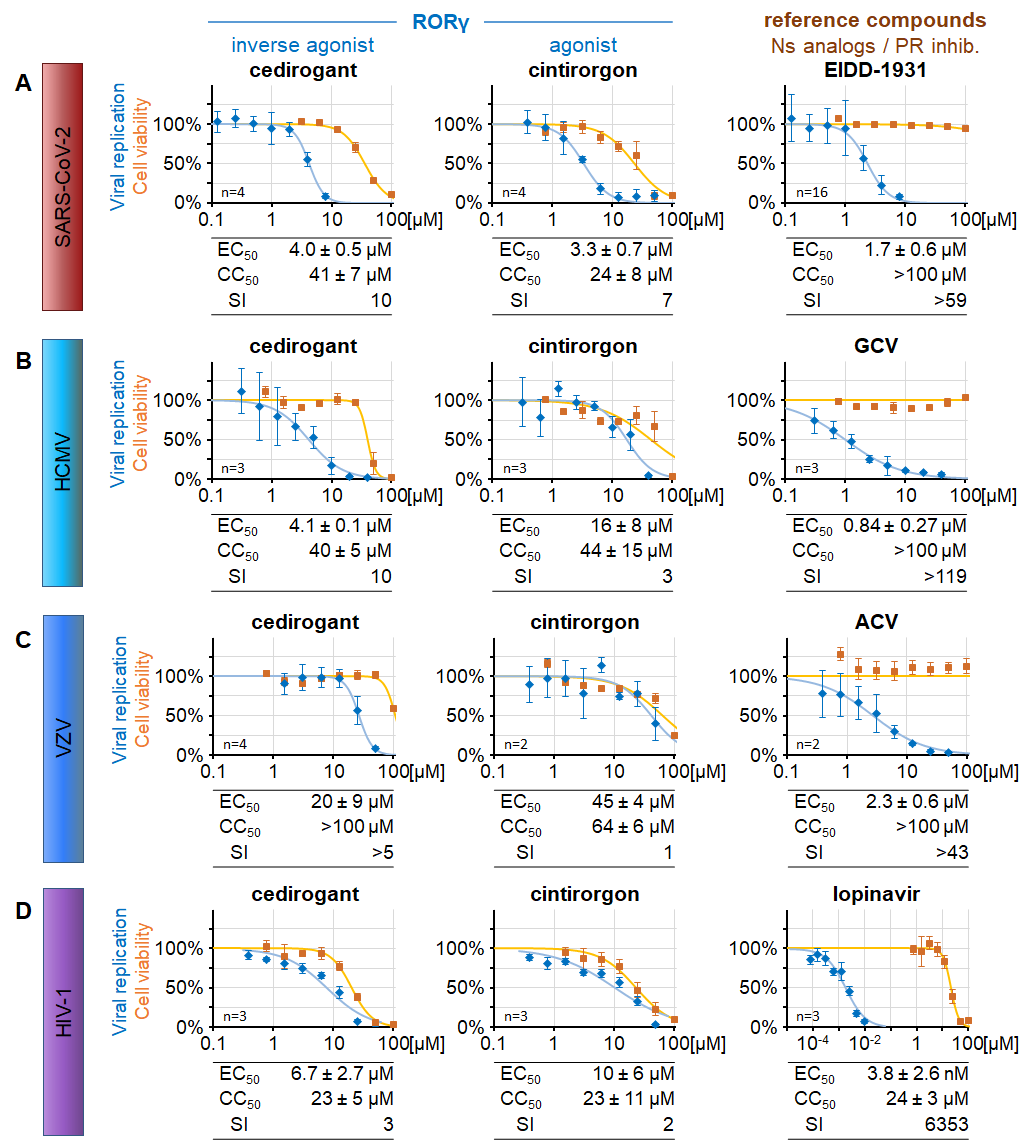
(A) Caco-2 cells infected with SARS-CoV-2 d6-YFP at MOI 0.003, (B) HFFs infected with HCMV AD169-GFP at MOI 0.001, (C) HFFs co-cultured with VZV Oka-GFP-infected cells (ratio 40:1), (D) or CEMx-GFP cells infected with HIV-1 NL4-3 at MOI 0.001 were treated with descending concentrations of the RORγ modulators cedirogant or cintirorgon or an appropriate reference compound. Viral replication was measured by quantitation of the respective viral reporter signal. Cell viability was determined in parallel with uninfected cell cultures (Neutral Red assay) for the equivalent duration of exposure to the compounds. For details, see Wangen, Raithel et al., 2023.
Host CDKs as promising targets for anti-HCMV and related antiviral treatment strategies
On an even broader scale, we demonstrated the importance of additional host CDKs for HCMV replication, including CDK2, CDK4/6, CDK9 and, very recently, CDK8. In particular, CDK8 ranged into the focus of our specific interest and might represent another highly interesting drug target. This is particularly true when considering the modern options for chemical modification of CDK target binders, i.e. through the coupling of a degrader unit. This strategy is based on the use of 'Proteolysis Targeting Chimeras' (PROTACs), which are hetero-bifunctional molecules containing two small molecule-binding ligands joined together by a linker. Such chemical linkage of PROTAC moieties is a means of recruiting a specific E3 ligase, thereby inducing the proteasome-mediated degradation of the target protein. Such PROTAC approaches, which are intended to enhance antiviral drug activity compared to their parental, non-degrading inhibitors, are especially worked out in the context of CDK targeting, so that the generation of an efficient CDK8-PROTAC is being intensely pursued. Thus, the longer-term objectives of this research line are the assessment of broadly acting antiherpesviral prototype compounds and the specific nomination of candidate drugs or combinatorial drug synergies. Beyond that, the use of CDK inhibitors in our projects provides a powerful tool for the analysis of mutual interregulation between HCMV and the host CDK–cyclin machinery.
Very similar approaches of drug targeting are now also developed for additional herpesviruses, such as Epstein-Barr virus (EBV), human herpesvirus 6 (HHV-6), rhesus macaque cytomegalovirus (RhCMV), and for a panel of non-herpesviruses. For these virus systems, specific reporter modules have been established, which now allow for the analysis of regulatory details of virus replication in host cell types and for the assessment of antiviral candidate drugs.
Concerning our comparative analyses with non-herpesviruses, we explored the human nuclear receptor and transcription factor RORγ isoform 1 (RORγ1), a member of the retinoic acid receptor-related orphan receptor (ROR) family, as a putative target of antiviral HDAs. To this end, cell culture models were used to investigate major viral human pathogens, i.e. the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), human cytomegalovirus (HCMV), varicella zoster virus (VZV) and human immunodeficiency virus 1 (HIV-1). Our results demonstrated (i) an antiviral activity of the clinically relevant RORγ modulators cedirogant and others, (ii) that isoform RORγ1 acts as the responsible determinant and drug target in the analyzed cell culture-based models, (iii) a selectivity of the antiviral effect for RORγ1 over related receptors RORα and RORβ, (iv) a late-phase inhibition exerted by cedirogant in HCMV replication and (v) a mechanistic link to the cellular cholesterol biosynthesis (Figure 9). Combined, the data highlight this novel RORγ-specific antiviral targeting concept and the developmental potential of RORγ-directed small molecules. For future studies from 2024 on, this investigation of the RORγ role in virus infections will be further developed by Friedrich Hahn as an independent research line.

