
Human Herpesvirus 8 (Kaposi's Sarcoma-associated Herpesvirus)
Since the beginning of the AIDS epidemic, three human herpesviruses have been discovered. DNA fragments of human herpesvirus 8 (HHV-8), also termed Kaposi sarcoma-associated herpesvirus (KSHV) were first identified in 1994 in AIDS associated Kaposi’s sarcoma biopsy specimens by representational difference analysis. DNA of this virus is invariably found in Kaposi’s sarcoma, body cavity based lymphomas, and certain forms of Castleman’s disease.
Herpesvirus entry into host cells
Entry of herpesviruses into host cells is seen as a multistep process involving several cellular receptors and at least four viral envelope glycoproteins of which only three (glycoproteins H, L and B) are conserved amongst all herpesviruses. The first step is attachment of the virion to the cytoplasma membrane. In most human herpesviruses, this step is mediated by one or more of the virus- or genus-specific glycoprotein(s) which bind to specific cellular receptor(s) and are often responsible for the cell tropism of the respective herpesvirus. In KSHV glycoprotein gpK8.1 is important for attachment of the virion to the cell by binding to heparansulfate. These strain-specific viral glycoproteins form complexes with the highly conserved glycoproteins H and L (gH/gL), either following receptor binding or already before, and seem to ‘activate’ gH/gL which – at least in some herpesviruses – is followed by endocytotic uptake of the virion. Interaction of activated gH/gL with glycoprotein B (gB) is then required to trigger the last step in herpesvirus entry: Fusion of the virion envelope with cellular membranes. This step is executed by trimeric gB which shares structural similarities with both class I and class II fusion proteins. As described above, a complex formed by the conserved glycoproteins gH/gL and at least one virus-specific receptor-binding protein is required to trigger gB mediated fusion in most herpesviruses.
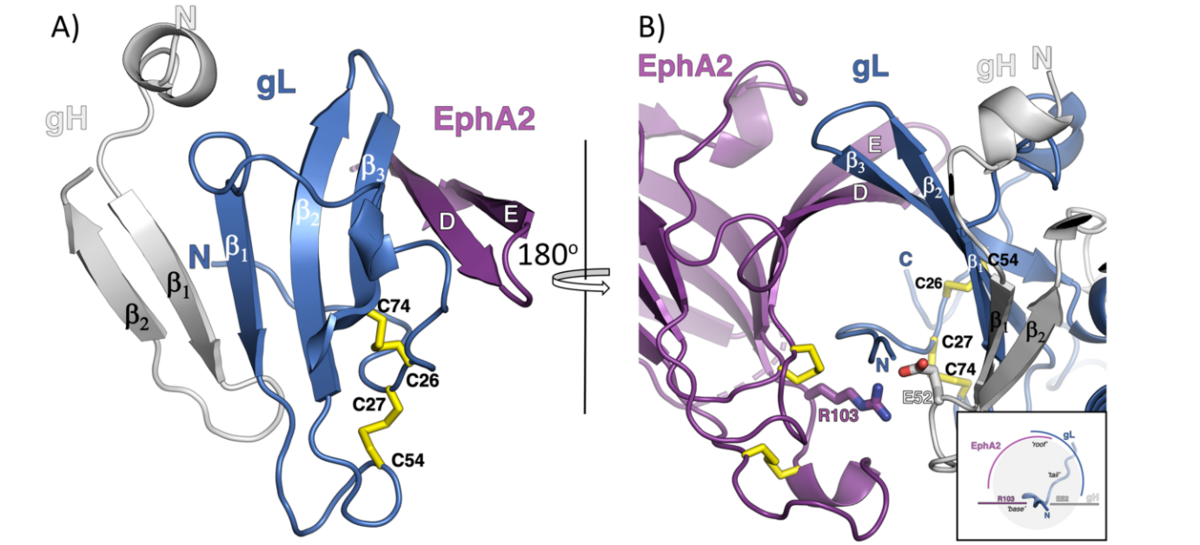
A) The mixed ß-sheet formed by strands of gH, gL and EphA2. The strands in gH and gL are labeled as ß number, while the EphA2 LBD strands are marked using the nomenclature assigned for the first solved structure of the EphB2.
B) View are the gH/gL and EphA2 binding interface from the other side with the inlet illustrating the channel formed by the EphA2 and gL strands (‘roof’) that hosts the gL N-terminal ‘tail’, reinforced by polar interactions between R103 on EphA2 and E52 on gH (‘base’).
Figure 1 is modified from T. P. Light et al., Plos. Biol. 2021.
EphrinA2 receptor-tyrosinkinase (EphA2) is involved in KSHV entry via endocytosis
Using immunoprecipitation and mass spectrometry we identified EphA2 as a cellular binding partner of KSHV gH/gL in 2012. We showed that EphA2 does not only bind to KSHV gH/gL with high affinity and specificity but that EphA2 is important for the infection of endothelial and epithelial cells by KSHV. Surprisingly, neither the intracellular domain nor the extracellular fibronectin domains of EphA2 were found to be required for KSHV infection of epithelial cells. In collaboration with Maria Backovic from the Institute Pasteur in Paris (group of Felix Rey) we analysed the interaction of the extracellular domain of EphA2 with the gH/gL complex of KSHV. Using X-ray structure analysis, targeted mutagenesis and binding studies, we here show that the HHV-8 envelope glycoprotein complex gH/gL binds with sub-nanomolar affinity to EphA2 via molecular mimicry of the receptor's cellular ligands, ephrins, revealing a pivotal role for the conserved gH residue E52 and the amino-terminal peptide of gL (figure 1).
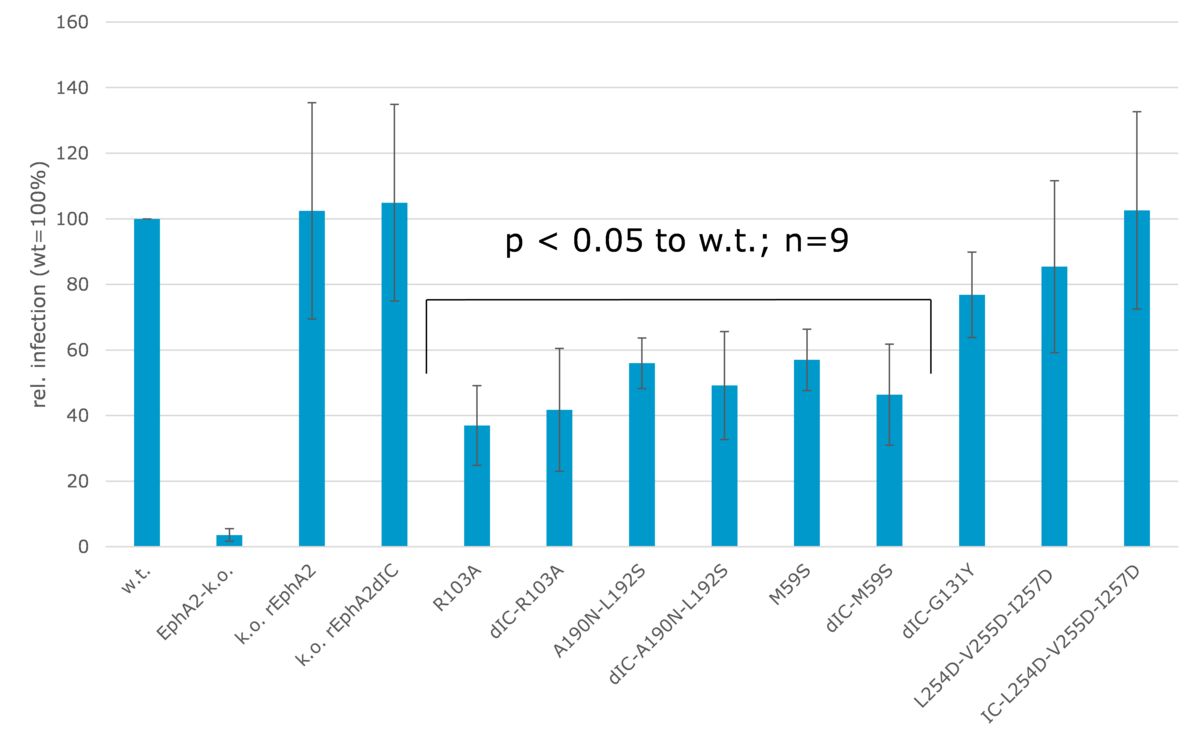
EphA2-k.o. cells were transfected with expression vectors for the indicated EphA2-mutants via retroviral vector. This was followed by infection with the recombinant GFP-expressing variant rKSHV.219. Infection rates were determined by flow cytometry.
We were next able to confirm at least in part the functional relevance of these interactions by creating mutants of EphA2 which were expressed in EphA2 knock-out cells (figure 2). Structure-driven mutants of EphA2 residues at the binding interface as well as the dimerisation and clustering interface were constructed and expressed in EphA2 knock-out cells. This confirmed that the direct interaction of gH/gL with EphA2 is important for infection, as mutants in the ligand binding domain (R103A, A190N – L192S, M95S) significantly reduced infection rates (figure 2). However, although dimerization is triggered by gH/gL binding and required for the induction of signal transduction, a mutation inhbiting dimerization did not have a significant influence on cell infectability (figure 2, dIC-G131Y). This is consistent with our finding that the intracellular domain of EphA2 is not required to trigger KSHV endocytosis.
Identification of antibodies blocking KSHV infection
Almost all tumor cells of KSHV-associated malignancies harbor the viral genome in a latent state. However, the development of KS-lesions is associated with an increase of antibodies against lytic KSHV-proteins indicating that lytic replication precedes KS development. Nevertheless, antiviral drugs currently available against KSHV and other herpesviruses are not effective against KSHV-associated malignancies. Passive and/or active immunization against KSHV would thus be important for people at risk for KS due to immunosuppression or living in areas of high KSHV prevalence. group of Frank Neipel studies function and immunogenicity of KSHV glycoproteins since the discovery of the virus. We identified the KSHV-specific glycoprotein gpK8.1 and were able to show that it mediates virion attachment by binding to heparansulfate. In addition, as described above, we identified two cellular receptors for the glycoprotein complex gH/gL and were able to show that soluble variants of the cellular protein EphA2 efficiently neutralize KSHV in cell culture. Neutralizing activity is detectable in the sera of KSHV infected individuals. However, the targets of these neutralizing antibodies have not been clearly identified, according to our data gpK8.1 and gH/gL are very likely candidates. We thus analysed the neutralising antibody targets in human sera.
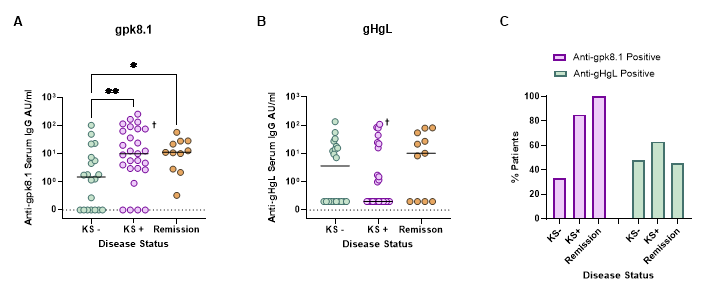
gpk8.1- and gHgL-specific ELISAs were used to measure the levels of anti-gpk8.1 (A) and anti-gHgL (B) antibodies in patient serum samples from those with asymptomatic infection (KS-), active disease (KS+) and from those in remission from KS (Remission) (AU: arbitrary units). Values were plotted on scatter plots and the median antibody level displayed for each disease group. gpk8.1 and gHgL responses were then compared as a percentage of patients with positive or negative ELISA responses (C) (Abs: antibodies). † refers to a patient who was in partial remission from KS. Data was analysed using Kruskal-Wallis tests (A: p = 0.014, B: p = not significant), followed by uncorrected Dunn’s test for multiple comparisons (* = p ≤ 0.05, ** = p ≤ 0.01). Uncorrected tests were used for multiple comparisons due to the small sample size in the remission group which may otherwise lead to type II error.
Remission from KS is associated with an increased gpk8.1 antibody response
The levels of gpk8.1- and gHgL-specific antibodies from patients with asymptomatic HHV-8 infection, active disease and those in remission from KS were measured via gpk8.1- and gHgL-specific ELISAs. The antibody levels were then compared between the different disease groups (Figure 3). The level of gpk8.1 binding antibodies was significantly raised in the sera of patients experiencing active disease and in those in remission from KS compared to sera from asymptomatic infected patients (Figure 3A). No significant difference was observed between the levels of gHgL binding antibodies and the different disease status groups (Figure 3B). In order to better visualise the pattern in the number of patients with a positive gpk8.1- or gHgL-specific ELISA response, the percentages of ELISA positive patients were determined for both anti-gpk8.1 and anti-gHgL responses (Figure 3C). The percentage of patients with antibodies against gpk8.1 was increased in those with active disease (KS+: 85 %) compared to those with asymptomatic infection (KS-: 33 %). Notably, all serum samples taken from those in remission were positive for gpk8.1 antibodies. This increase in the number of gpk8.1 positive responders during disease progression was not reflected by the gHgL positive responders.
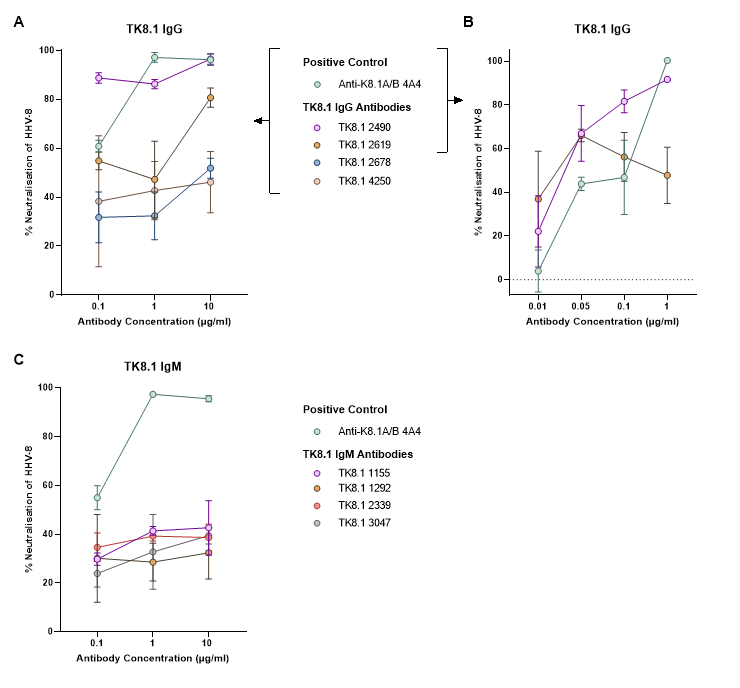
Purified gpk8.1-specific monoclonal antibodies (TK8.1) were incubated at a range of dilutions with rKSHV.219 before addition to MC116 cells. The GFP expression was then quantified via flow cytometry as a measure of infection. Along with the positive control (anti-K8.1A/B 4A4), two IgG TK8.1 antibodies (TK8.1 2490 and TK8.1 2619) were identified as being able to block HHV-8 infection (A). This neutralising effect was present down to 0.05 µg/ml (B). Results were normalised to the non-specific TRES 224 IgG antibody (A and B). The neutralising activity of the TK8.1 IgM (C) antibodies were also assessed and normalised to a non-specific isotype control (anti-nuclear spliceosomes IgMκ). The mean and standard deviation of three replicates are displayed.
Antibodies against the N-terminal domain of gpK8.1 block KSHV infection
A vaccination platform targeting gpk8.1 was tested using multimeric gpk8.1 in an attempt to enhance antigen immunogenicity. Monomeric, dimeric and trimeric gpk8.1, using Fc and T4 foldon domains for dimerisation and trimerisation respectively, were produced and used in mouse immunisations. Immunisation was found to induce gpk8.1-specific antibodies, and in most cases could induce an HHV-8 neutralising response. In an effort to isolate a neutralising antibody, gpk8.1-specific monoclonal antibodies were produced from trimeric gpk8.1 immunised TRIANNI mice. Two of these monoclonal antibodies were able to block HHV-8 infection in the B cell line MC116, which lacks HS on its surface (figure 4). This is in support of previously published data and suggests that gpk8.1 is essential for B cell infection, independent of its known HS binding properties. The interaction of these neutralising antibodies with gpk8.1 to an unknown receptor on B cells was localised to the first 24 amino acids of gpk8.1, indicating the likely location of a binding domain.

