
Comparative Viral Genetics
General background
The worldwide Covid-19 (Corona VirusDisease-19) pandemic between 2019 and 2023 has opened unprecedented challenges for research on viral genetics. The causative agent of the disease, the newly emerged SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus-2), has been intensely studied under many different aspects by researchers worldwide. Our group initially became interested to follow the emergence of new mutants and variants [1-3]. In the course of this more systematic work, the idea came up to compare the spike nucleotide sequence of SARS-CoV-2 with the spike sequences of human Adenoviruses2 and 12 and eventually of many different species, from viruses to Homo sapiens [4-7].
Starting in 1966, human Adenoviruses have been a major research tool for investigations in mammalian molecular genetics also in Walter Doerfler’s laboratory. Our work was started in 1966 at the Rockefeller University in New York, N.Y., continued starting in 1972 at the Institute for Genetics at Cologne University and, since 2002, at the Institute for Clinical and Molecular Virology, Friedrich-Alexander-University Erlangen-Nürnberg. Integrative recombination, the structure of junction sites with integrated foreign DNA, inverse correlations between DNA methylation and transcriptional activity as well as genome-wide alterations of methylation patterns that followed foreign DNA insertions into mammalian genomes have been topics of our long-standing interest [summary in references 4-7].
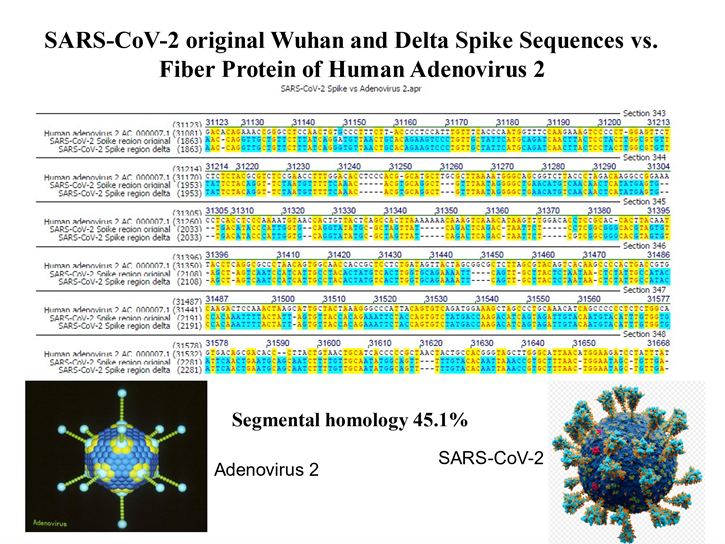
Driven by curiosity, we compared the full lengths spike nucleotide sequences between SARS-CoV-2 and Human Adenoviruses and made the serendipitous discovery of ~ 45% sequence identities between these viral genomes. A more detailed inspection of the identity profiles revealed patch type sequence identity patterns similar to those we and many others had found at the junction sites of illegitimate recombination partners, among others at the junction sites of Adenovirus DNA integrates into mammalian genomes [summary in references 4, 5]. Extension of the sequence alignments to the genomes of completely different species – as far apart as human and Oryza sativa (rice) or SARS-CoV-2 and Ilex aquifolium - invariably revealed ~ 45% patch type sequence identities, often along the entire lengths of the alignments with the shorter partners. Here, we entered the field of Comparative Viral Genetics with important consequence for biology and medicine [4-7]. We have argued that these unexpectedly high levels of inter-species nucleotide sequence identities might be an inherent built-in feature of a four-letter genetic alphabet. This notion is supported by the finding in control experiments that among 1.000 artificial genomes generated by shuffling A, C, G, and T, patchy sequence identities have been invariably observed [4-6].
Our research between 2019 and 2024 was facilitated by the free availability of large amounts of nucleotide sequence data from sources like GISAID and the NCBI nucleotide databases.
Between 2019 and 2023, our research group has concentrated on the following topics:
- The nature and possible biological significance of patch type identities between the genomes of distantly or unrelated species and SARS-CoV-2 evolution
- The rapid worldwide rise and selection of SARS-CoV-2 mutants and variants
A built-in feature of the four-letter genetic alphabet – patchy sequence identities between genomes of diverse species are key to illegitimate recombination and evolution
Patch-type nucleotide sequence identities were detected among genomes of a wide and diverse spectrum of biological taxa, including viruses, bacteria, plants, animals, and humans. Surprisingly, a comparison of the genomes of Adenoviruses and SARS-CoV-2 revealed patch type sequence identities of ~ 45% along the entire lengths of the global viral genome alignments. Extending sequence comparisons to 65 genome alignments of distantly or unrelated species, the same patterns of patchy sequence identities at ~ 45% recurred. In silico control experiments showed similar percent identities with patchy patterns when A, C, G, and T sequences were randomly permuted. A four-letter genetic alphabet and its possibly built-in ability to give rise to patch type identity patterns with unrelated sequences facilitate genetic exchange and recombination processes. This latter conclusion is based on results from the early 1980s of patchy sequence patterns at junction sites of recombination or integration events in many biological systems. The present study also explored the potential role of patchy identity patterns to cause the high rates of mutation in SARS-CoV-2 and its Omicron variants. In the Omicron variant BA.2.86, a 22-nucleotide exchange was documented between the genomes of the original SARS-CoV-2 Wuhan-Hu-1 reference genome and the BA.2.86 Omicron variant. In this exchange reaction, patchy identity patterns were also observed. Moreover, individual mutations in the viral spike gene are often only a few nucleotides apart. Hence, patch-type sequence identities may have served as specific signals for illegitimate recombination.
Background and Discovery: Emanating from the chance observation of ~ 45% patchy sequence identities between the spike sequences of SARS-CoV-2 and Human Adenoviruses, we have detected the same ~ 45% sequence identities in genome comparisons of many diverse species, e.g., Homo sapiens versus Ilex aquifolium. So far, these identities have been confirmed in 65 sequence alignments of genomes from very different species [4-5]. The patch-type sequence identities can be fully appreciated only when one scrolls through the full lengths of aligned sequences, e.g. of the Adenovirus and SARS-CoV-2 genomes [see supplemental material of references 4, 5].
Bioinformatics Controls: Similar percent identities were observed when the sequence of each organism was randomly permuted and then the alignments were re-done using the same parameters. Moreover, the analyses were repeated among 1,000 alignments using 40 permutations of parameters in the publicly available global blast alignment. The percent identities could vary around 10% depending on the choice of parameters. In all of the experiments, patch type identities were found that were dispersed throughout the entire lengths of the alignments. These analyses were performed by Ch. Ramirez, Professor of Biostatistics at UCLA, Los Angeles. Our preliminary interpretation of this remarkable finding suggests a presumptive built-in property in the four-letter genetic alphabet.
Interpretation:Conclusions in the light of our earlier studies from the 1980’s and 1990’s [see references 4-6] on patchy nucleotide sequence homologies/identities at junction sites between Adenovirus 12 DNA integrates and mammalian cellular DNAs recall that such structures probably serve as signals for illegitimate recombination, sequence exchange or integration reactions in mammalian cells.
Possible relevance for an understanding of the origin of SARS-CoV-2 and its continuously emerging Omicron variants: The nucleotide sequences of Omicron variants BA.1, BA.2, BA.2.75, BA.4, BA.5 and XBB.1.5 of SARS-CoV-2 were inspected for distances between neighbouring mutations that proved to be very closely spaced, in many cases only a few nucleotides apart [5]. Close mutation spacing was particularly noted in the receptor domain of the spike segment of these SARS-CoV-2 genomes [4, 5]. Even more to the point, in the Omicron variant BA.2.86 („Pirola“), a 22-nucleotide exchange was documented between the genomes of the original SARS-CoV-2 Wuhan-Hu-1 reference genome and the BA.2.86 Omicron variant. In this 22-nucleotide sequence exchange segment, 2 four- and 2 two-patch type identities were detected. This finding suggests a direct sequence exchange between SARS-CoV-2 RNA and unknown, possibly cellular, RNAs.
Open Questions and Transient Therapy: In the future, we will pursue the biochemical and genetic mechanisms of illegitimate recombination in mammalian cells. Therapeutic considerations for viral infections raise the question of whether this mechanism could be interfered with or even be temporarily halted as a transient therapeutic measure during a viral pandemic? Moreover, we plan to extend the number of sequence alignments, since our to-date repertoire of investigated sequence comparisons is still limited.
Conclusions: Patch-type nucleotide sequence identities were observed among the genomes of various and even unrelated species. These identities could allow for inter-molecular sequence exchanges by a mechanism akin to heterologous or illegitimate recombination reactions. The results of computational analyses, including randomly permutated sequences generated from the A, C, G, T/U 4-letter alphabet, revealed that the 43% to 50% sequence identities and their patchy patterns were maintained. The patchy identity patterns might be related to a built-in property of the four-letter alphabet that must have been selected very early-on in evolution since it pervades all forms of life.
In many integration or illegitimate recombination reactions, the sites of linkage between foreign and host DNAs, are characterized by patchy sequence identities, by partial deletions or insertions of short sequences of unknown derivation [4-5]. These frequently recurring observations in very different biological systems imply that sequence identity-based stem-loop structures have the potential to serve as signals in the recognition between mutual recombination partners.
A sizable literature exists that describe recombination between RNA molecules, among the genomes of distantly related viral RNAs and even with any RNA that is available to serve as recombination partner [reviewed in references 4, 5].
The distribution patterns of mutations in the glycoprotein spike gene of SARS-CoV-2 are characterized by closely spaced point mutations by short deletions and insertions, hence might be due to exchanges between short sequence stretches, e. g., from the human host genome or human/viral RNAs. The rapid appearance of new variants of SARS-CoV-2, particularly of the Omicron family, renders this hypothesis worth considering. More specifically, comparison of the nucleotide sequences of the SARS-CoV-2 Wuhan Hu-2 reference and the SARS-CoV-2 Omicron variant BA.2.86 (Pirola) reveals four patch type sequence identities involving two of 2 and two of 4 nucleotides in lengths.
These observations underscore the significance of patchy sequence identities in the context of sequence exchange and recombination processes. However, there still are limitations to this study and further research will be needed to elucidate the nature of unknown evolutionary events and the mechanisms governing illegitimate recombination and sequence exchange reactions.
The rapid worldwide rise and selection of SARS-CoV-2 mutants
Scientists and the public were alarmed by the occurrence of the large number of viral variants of SARS-CoV-2 reported starting in 2020. We have followed the time course of emerging viral mutants and variants during the SARS-CoV-2 pandemic in ten countries on four continents. We examined > 383,500 complete SARS-CoV-2 nucleotide sequences in GISAID (Global Initiative of Sharing All Influenza Data) with sampling dates extending until April 05, 2021. These sequences originated from ten different countries: United Kingdom, South Africa, Brazil, United States, India, Russia, France, Spain, Germany, and China. Among the 77 to 100 novel mutations, some previously reported mutations waned and some of them increased in prevalence over time. VUI2012/01 (B.1.1.7) and 501Y.V2 (B.1.351), the so-called UK and South Africa variants, respectively, and two variants from Brazil, 484K.V2, now called P.1 and P.2, increased in prevalence. Despite lockdowns, worldwide active replication in genetically and socio-economically diverse populations facilitated selection of new mutations. Rapidly evolving new variant and mutant strains might give rise to escape variants, capable of limiting the efficacy of vaccines, therapies, and diagnostic tests.
References
- [1] Weber S, Ramirez C, Doerfler W. Signal hotspot mutations in SARS-CoV-2 genomes evolve as the virus spreads and actively replicates in different parts of the world. Virus Research 2020 Nov; 289: 198170. doi: 10.1016/j.virusres.2020.198170.
- [2] Weber S, Ramirez CM, Weiser B, Burger H, Doerfler W. SARS-CoV-2 worldwide replication drives rapid rise and selection of mutations across the viral genome: a time-course study - potential challenge for vaccines and therapies. EMBO Mol Med. 2021, 13(6): e14062. doi: 10.15252/emmm.202114062.
- [3] Doerfler W. Adenoviral vector DNA- and SARS-CoV-2 mRNA-based Covid-19 vaccines: Possible Integration into the Human Genome - Are Adenoviral genes expressed in vector-based vaccines? Virus Research 2021 doi: 10.1016/j.virusres.2021.198466.
- [4] Weber S, Ramirez CM, Doerfler W. Ubiquitous micro-modular homologies among genomes from viruses to bacteria to human mitochondrial DNA: Platforms for Recombination during Evolution? Viruses. 2022 Apr 24;14(5):885. doi: 10.3390/v14050885.
- [5] S. Weber, C. Ramirez, W. Doerfler. A built-in feature of the four-letter genetic alphabet - Patchy sequence identities between genomes of diverse species are key to illegitimate recombination and evolution. Under revision, 2024 (manuscript available on request).
- [6] Doerfler W. Essential concepts are missing: Foreign DNA in food invades the organisms' cells and can lead to stochastic epigenetic alterations with a wide range of possible pathogenetic consequences. Clinical Epigenetics. 2020 Feb 7;12(1):21. doi: 10.1186/s13148-020-0813-z.
- [7] Doerfler W. Genome-wide Epigenetic Profiles (The Sixth Weissenburg Symposium) - Bericht. In: Jahrbuch 2021 der Deutschen Akademie der Naturforscher Leopoldina, Nationale Akademie der Wissenschaften; pages 93 - 110.

